The FDA implemented the breakthrough device program in 2015. The program was designed to expedite review of premarket approval (PMA), 510(k) clearance, and De Novo marketing authorizations. Applicants can seek a breakthrough device designation prior to requesting market approval regardless of the classification of the device. In order to qualify as a breakthrough device, the device must (1) provide for a more effective treatment or diagnosis of a life-threatening or irreversibly debilitating disease or condition and (2) meet at least one of: (a) represents a breakthrough technology; (b) no approved or cleared alternatives exist, (c) offers significant advantages over existing approved or cleared alternatives, or (d) device availability is in the best interest of patients.
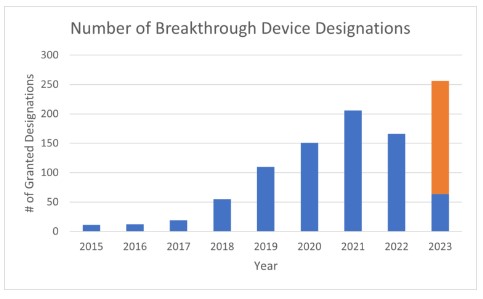
During the first year of breakthrough device program in 2015, the FDA granted only 11 breakthrough device designations. That number grew each year, peaking in 2021 with 206 designations. In 2022, the number of breakthrough device designations fell to 166, causing some to advocate for changes to the program. However, based on the most recent data from March 31, 2023, the FDA is on pace to grant expedited review to more than 250 devices in 2023. This would be an all time high.
The chart below illustrates the number of breakthrough device designations from 2015 through 2022, and extrapolates FDA’s pace of breakthrough device designations from the first quarter of 2023 through a full year.
The increase of breakthrough device designations has allowed more products to get to the commercial market in a shorter amount of time. Under the breakthrough device program, the average time for review of a 510(k) clearance decreased by more than 50%, down from 270 days to 155 days. Similarly, the breakthrough device program has decreased time to grant for a De Novo marketing authorization by 68 days, a decrease of more than 25%.
The decrease in time-to-approval under the breakthrough device program has sparked debate about the safety of approved breakthrough devices and benefit of the breakthrough device program. Patient-safety advocates argue that “with the breakthrough designation, the standard for evidence behind these devices has lessened.” Others maintain that the risk is justified and argue that patients are also harmed by delays in the approval process. Harvard Bioethics Professor Jonathan Darrow remains on the fence, suggesting that more post-market evidence of breakthrough devices is needed before the program can be properly assessed.
With the FDA continuing to grant larger numbers of breakthrough device designations, the breakthrough device designation may prove to be a worthwhile and cost-effective option for medical device companies that are seeking to reach the market faster with a device that meets the qualifications for the program.